Guide pratique : Syndrome CDK13
Auteurs : Flavien ROUXEL, interne de génétique et Pr David GENEVIEVE Centre de Référence Anomalies du Développement CHRU MONTPELLIER
Publié juillet 2020
OMIM #617360 ; ORPHA : non encore déterminé
Qu'est ce que le syndrome CDK13 ?
Le syndrome lié à CDK13, ou CHFIDD est un syndrome de découverte récente, identifié en 2016, grâce à l’amélioration des techniques de diagnostic en génétique. Il est dû à une anomalie au niveau d’un gène appelé CDK13, situé au niveau du bras court du chromosome 7 en situation 7p14.1
C’est un syndrome d’expression autosomique dominante, ce qui veut dire qu’il est lié à une atteinte d’un autosome, et qu’il suffit d’un exemplaire muté du gène pour être malade (on parle alors d’hétérozygotie).
Il entraine principalement des troubles des apprentissages ou une déficience intellectuelle, des difficultés à la nutrition tels qu’un reflux gastro-œsophagien dans l’enfance, des malformations cardiaques, une surdité et des crises d’épilepsie.

Caractéristiques cliniques
Le syndrome lié au gène CDK13 est un syndrome rare, mais le nombre de diagnostics est en augmentation grâce à la généralisation d’examens comme l’exome ou le génome, il faut donc noter que la description clinique donnée ci-après représente les personnes déjà décrites dans la littérature, mais que celle-ci peut évoluer avec l’ajout de nouvelles connaissances sur le syndrome lié à CDK13.
Ce syndrome associe :
Sur le plan psychosocial et cognitif :
Des troubles des apprentissages avec une déficience intellectuelle légère à modérée chez 95% des personnes porteuses du syndrome génétique lié à CDK13.
Un retard d’acquisition du langage verbal avec langage restreint ou absent. Les enfants atteints du syndrome lié à
Des troubles du spectre autistique ont été rapportés chez 26% des personnes atteintes.
Un trouble de déficit attentionnel ou une hyperactivité seule ont été rapportés dans 16.7% des cas.
Un PICA (ingestion de substances non nutritives et non comestibles) a été constaté chez 5% des personnes.
Sur le plan neurologique :
Les enfants atteints du syndrome lié à
Des crises d’épilepsie ont été constatées chez 19% des personnes, incluant crise myoclonique, crise tonico-clonique généralisée, absence.
Aucune corrélation entre anomalie de morphologie cérébrale et crises d’épilepsie n’a été mise en évidence.
Sur le plan ophtalmologique :
Un strabisme a été identifié chez 55% des personnes atteintes. Il ne semble pas y avoir d’autres anomalies oculaires fréquentes.
Sur le plan cardiaque :
Au total, 46% des enfants atteints de syndrome lié à
Ont aussi été rapportées des anomalies des artères pulmonaires chez 16% des personnes, une anomalie d’Ebstein et une tétralogie de Fallot chez 2.7% des personnes.
Sur le plan gastro-entérologique :
La plupart des enfants atteints de syndrome lié à CDK13 ont des antécédents de difficultés pour se nourrir, de trouble de l’oralité, avec par exemple une lenteur aux repas ou un reflux gastro-œsophagien, et 16% des personnes nécessiteront une alimentation par sonde de gastrostomie.
Environ 12% des enfants atteints de syndrome lié à CDK13 ont également une constipation sévère.
Sur le plan musculosquelettique :
Les anomalies spinales comportent : scoliose, hyperlordose, hémangiomes vertébraux, fusions de vertèbres cervicales, spina bifida, proéminence du sacrum.
Des contractures musculosquelettiques ont été décrites dans 12.5% de cas, présumées secondaires à la spasticité. Elles consistent en une contracture des extenseurs du cou et de la colonne vertébrale limitant la flexion du cou et de la colonne vertébrale cervicale, et contribuent à une posture atypique en hyperextension apparue dans la première année de vie.
Sur le plan dentaire :
Des dents espacées en forme de cône ont été rapportées chez un peu moins de 10% des personnes atteintes de syndrome lié à CDK13. Il est nécessaire d’évaluer la mise en place de la denture définitive en particulier s’il existe un palais creux.
Sur le plan néphrologique :
Les anomalies rapportées sont des duplications ou dilatations des canaux collecteurs, ou un rein en fer à cheval ou ectopique. La prévalence exacte des anomalies rénales n’est pas connue par manque d’imagerie rénale dans les cas rapportés de syndrome lié à CDK13.
A noter qu’1/3 des personnes atteintes de syndrome lié à
Pronostic :
La sévérité de la pathologie est variable, et nous disposons de peu de recul sur cette pathologie, aussi, il n’est pour le moment pas possible de présumer de l’évolution clinique des personnes.
Aspects génétiques
Nous possédons tous 46 chromosomes, répartis en 23 paires avec, pour chaque paire, un chromosome hérité de notre mère et un chromosome hérité de notre père. Parmi ces 23 paires de chromosomes, on distingue les autosomes (chromosomes 1 à 22) qui sont communs aux filles et aux garçons, et les gonosomes qui sont les chromosomes sexuels : XX chez les filles et XY chez les garçons. Chacun de ces chromosomes porte des centaines de gènes, qui permettent le bon fonctionnement de l’organisme grâce à la production de protéines.
Dans ce syndrome, une des copies du gène CDK13 est atteinte. L’autre est normale, mais ne suffit pas à compenser cette perte. On parle d’expression autosomique dominante.
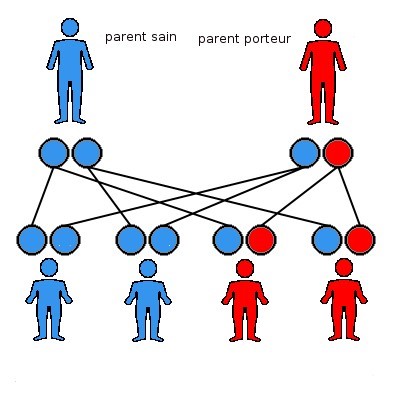
Lors de la conception d’un embryon, la maman donne 23 chromosomes, et le papa donne aussi 23 chromosomes, ce qui donne 4 combinaisons possibles de patrimoine génétique pour l’enfant, schématisées ci-dessus. Chaque rond représente un lot de chromosomes.
Dans le cadre d’une maladie autosomique dominante, le parent porteur a donc un risque sur deux de transmettre la mutation (ici schématisée en rouge). L’enfant a un risque sur deux de porter la mutation, et donc de présenter la maladie.
Diagnostic
Pour le moment, aucun critère clinique formel de diagnostic du syndrome lié à CDK13 n’a été décrit.
Le diagnostic repose actuellement sur l’identification à l’état hétérozygote d’un variant pathogène de CDK13 par analyse de génétique moléculaire. La pénétrance est complète, ce qui veut dire que la présence d’un variant pathogène dans le gène CDK13 entraine la présence de la maladie.
On peut arriver à un tel résultat :
- Soit en prescrivant un panel de gènes contenant le gène CDK13
- Soit en prescrivant une analyse plus générale, comme une analyse d’exome, c’est-à-dire une analyse de toutes les séquences codantes de tous les gènes connus en pathologie humaine, ou une analyse de génome, c’est-à-dire une analyse de toutes les bases de l’ADN.
Conseil génétique
Pour le moment, toutes les personnes porteuses de syndrome lié à CDK13 rapportées sont sporadiques, c’est-à-dire que les parents de l’enfant ne portent pas la mutation dans leur patrimoine génétique. Le variant pathogène dans le gène CDK13 provient d’un « accident » génétique qui se produit dans le spermatozoïde ou l’ovule. On parle alors de variant pathogène (anciennement mutation) de novo.
Il peut arriver que plusieurs spermatozoïdes ou plusieurs ovules portent le variant pathogène alors qu’il n’est présent nulle part ailleurs dans le corps des parents. Ce mécanisme s’appelle une mosaïque germinale.
Devant un risque de mosaïque germinale estimé à 1%, une consultation de génétique est possible dans le but de proposer un diagnostic prénatal aux parents d’un enfant atteint pour une future grossesse s’ils désirent s’assurer que le fœtus ne porte pas le variant pathogène.
Ce diagnostic prénatal n’est pas obligatoire et est à discuter avec la famille.
Prise en charge globale, traitement et suivi
Traitement :
Il n’existe à ce jour pas de traitement curatif du syndrome lié à CDK13.
On peut cependant prévenir ou traiter certains symptômes comme :
- Les crises d’épilepsie pour lesquelles on peut donner des antiépileptiques
- L’hyperactivité et les troubles du comportement pour lesquels on peut donner du méthylphénidate
- La constipation pour laquelle on peut donner des règles hygiénodiététiques et des laxatifs
- Les troubles de la vue pour lesquels on peut prescrire le port de lunettes
- Les anomalies dentaires pour lesquelles est indiquée une prise en charge orthodontique
De plus, il existe des moyens de prise en charge chirurgicale, notamment pour les malformations cardiaques ou les problèmes orthopédiques.
Surveillance et suivi:
Le syndrome lié à CDK13 nécessite un suivi spécialisé tout au long de la vie. Ce suivi sera coordonné par le médecin du centre de référence ou de compétence, puis par le pédiatre ou le médecin généraliste, et adapté en fonction des problèmes médicaux spécifiques de la personne.
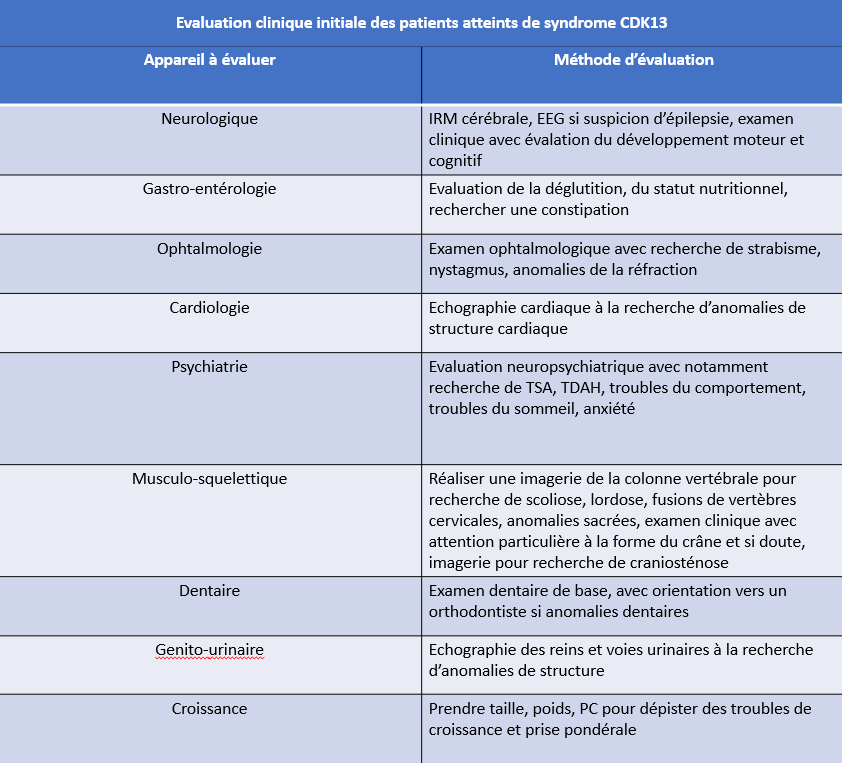
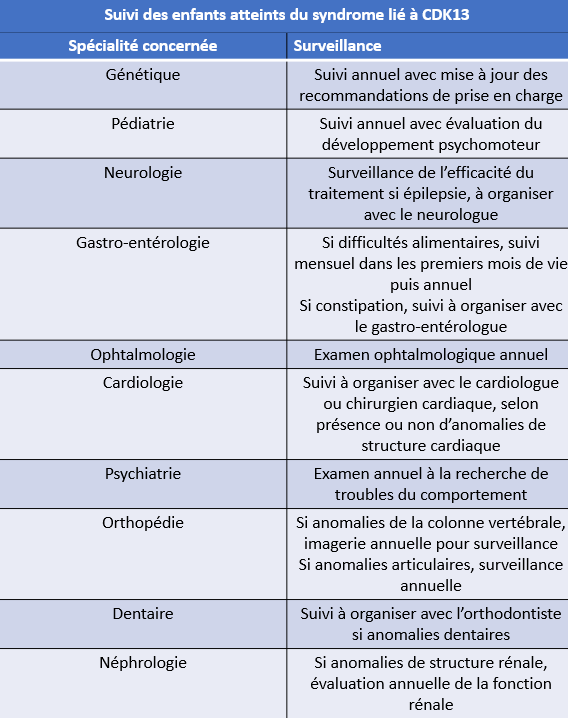
Scolarisation
La prise en charge du retard de développement et des difficultés d’apprentissage doit être la plus précoce possible. Il est important de favoriser une scolarisation en milieu ordinaire, avec, si besoin, mise en place d’aménagements, d’aides matérielles et d’aides humaines (AESH Accompagnant des Elèves en Situation de Handicap, anciennement appelé AVS). Un Projet Personnalisé de Scolarisation (PPS) doit être rédigé en lien avec les équipes éducatives. Dans certains cas, une orientation en milieu éducatif spécialisé est nécessaire.
Suivi paramédical
Des prises en charge rééducatives adaptées aux besoins de chaque personne doivent aussi être mises en place le plus précocement possible, telles que l’orthophonie, la psychomotricité, la kinésithérapie, le suivi par une psychologue, ... Les bilans réalisés par ces différents professionnels, et en particulier l’évaluation neuropsychologique, sont très importants puisqu’ils permettent d’identifier les points forts et les points faibles de chaque personne. Ainsi, les apprentissages scolaires seront facilités en s’appuyant sur les points forts et en compensant ou en contournant les difficultés liées aux faiblesses.
Accès aux droits
Des aides financières, telle que l’Allocation pour l’Education des Enfants en situation de Handicap (AEEH), peuvent également être mises en place pour compenser la prise en charge du handicap. L’AEEH est basée sur les constatations des professionnels médicaux et paramédicaux d’une part, et des aidants principaux d’autre part.
Le médecin référent se charge de remplir la demande de prise en charge à 100% au titre d’une Affection Longue Durée (ALD) hors liste. Il remplit également le certificat médical pour le dossier de la Maison Départementale de l’Autonomie (MDA) (anciennement appelée Maison Départementale des Personnes en situation de Handicap, MDPH).
Enfin, le réseau Maladies Rares Méditerranée est également à votre disposition pour vous accompagner dans les différentes démarches sociales : aide à la constitution du dossier pour la Maison Départementale de l’Autonomie (MDA), …
Associations de patients
Association Française :
Page facebook : Les jolis pas d'Eléa
@lesjolispasdelea · Communauté
Internationale :
Pages facebook CDK13 - Genetic Mutation
@CDK13Geneticmutation · Communauté
Références
1.Chen H-R, Lin G-T, Huang C-K, Fann M-J. Cdk12 and Cdk13 regulate axonal elongation through a common signaling pathway that modulates Cdk5 expression. Exp Neurol. nov 2014;261:10‑21.
2.Bostwick B. CDK13-Related Disorder. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al., éditeurs. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993 [cité 12 mars 2020].
Disponible sur: http://www.ncbi.nlm.nih.gov/books/NBK536784/
3.Hamilton MJ, Suri M. CDK13-related disorder. Adv Genet. 2019;103:163‑82.
4.Liang K, Gao X, Gilmore JM, Florens L, Washburn MP, Smith E, et al. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol Cell Biol. mars 2015;35(6):928‑38.
5.van den Akker WMR, Brummelman I, Martis LM, Timmermans RN, Pfundt R, Kleefstra T, et al. De novo variants in CDK13 associated with syndromic ID/DD: Molecular and clinical delineation of 15 individuals and a further review. Clin Genet. 2018;93(5):1000‑7.
6.Sifrim A, Hitz M-P, Wilsdon A, Breckpot J, Turki SHA, Thienpont B, et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet. 2016;48(9):1060‑5.
7.Hamilton MJ, Caswell RC, Canham N, Cole T, Firth HV, Foulds N, et al. Heterozygous mutations affecting the protein kinase domain of CDK13 cause a syndromic form of developmental delay and intellectual disability. J Med Genet. 2018;55(1):28‑38.
8.Greenleaf AL. Human CDK12 and CDK13, multi-tasking CTD kinases for the new millenium. Transcription. 2019;10(2):91‑110.
9.Nováková M, Hampl M, Vrábel D, Procházka J, Petrezselyová S, Procházková M, et al. Mouse Model of Congenital Heart Defects, Dysmorphic Facial Features and Intellectual Developmental Disorders as a Result of Non-functional CDK13. Front Cell Dev Biol. 2019;7:155.
10.Trinh J, Kandaswamy KK, Werber M, Weiss MER, Oprea G, Kishore S, et al. Novel pathogenic variants and multiple molecular diagnoses in neurodevelopmental disorders. J Neurodev Disord. 25 2019;11(1):11.
11.Yakubov R, Ayman A, Kremer AK, van den Akker M. One-month-old girl presenting with pseudohypoaldosteronism leading to the diagnosis of CDK13-related disorder: a case report and review of the literature. J Med Case Rep. 29 déc 2019;13(1):386.
12. Bostwick BL, McLean S, Posey JE, Streff HE, Gripp KW, Blesson A, et al. Phenotypic and molecular characterisation of CDK13-related congenital heart defects, dysmorphic facial features and intellectual developmental disorders. Genome Med. 14 2017;9(1):73.
13.Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 23 2017;542(7642):433‑8.4
14.Uehara T, Takenouchi T, Kosaki R, Kurosawa K, Mizuno S, Kosaki K. Redefining the phenotypic spectrum of de novo heterozygous CDK13 variants: Three personnes without cardiac defects. Eur J Med Genet. mai 2018;61(5):243‑7.
15.Greifenberg AK, Hönig D, Pilarova K, Düster R, Bartholomeeusen K, Bösken CA, et al. Structural and Functional Analysis of the Cdk13/Cyclin K Complex. Cell Rep. 12 janv 2016;14(2):320‑31.
Professionnels de santé en occitanie
Génétique médicale pour la coordination des soins - Centre de référence (constitutif) anomalies du développement et syndromes malformatifs sud-ouest, Occitanie, Réunion
Hôpital Arnaud de Villeneuve - CHRU de Montpellier
Pôle Biologie Pathologie - Département de Génétique Médicale
34295 Montpellier cedex 5
Cardiopédiatrie - Centre de compétence
Hôpital Arnaud de Villeneuve - CHRU de Montpellier
Pôle enfant - Département de pédiatrie
34295 Montpellier cedex 5
Cardiologie adulte pour le suivi à l'âge adulte - Centre de compétence
Hôpital Arnaud de Villeneuve - CHRU de Montpellier
Département de Cardiologie et maladies vasculaires
34295 Montpellier cedex 5
Chirurgie orthopédique et plastique infantile pour les scolioses - Centre de compétence
Hôpital Lapeyronie - CHRU de Montpellier
Pôle enfant - Département de chirurgie infantile
34295 Montpellier cedex 5
Endocrinologie pédiatrique pour les troubles hormonaux - Centre de compétence
Hôpital Arnaud de Villeneuve - CHRU de Montpellier
Pôle enfant - Département de pédiatrie
34295 Montpellier cedex 5
Gastrologie pédiatrique pour les difficultés alimentaires et troubles du transit - Centre de compétence
Hôpital Arnaud de Villeneuve - CHRU de Montpellier
Pôle enfant - Département de pédiatrie
34295 Montpellier cedex 5
Néphrologie pédiatrique pour les anomalies rénales - Centre de référence
Hôpital Arnaud de Villeneuve - CHRU de Montpellier
Pôle enfant - Département de pédiatrie
34295 Montpellier cedex 5
Neuropédiatrie et neurochirurgie pédiatrique pour le suivi développemental et les complications neurologiques - Centre de compétence
Hôpital Gui de Chauliac - CHRU de Montpellier
Département de pédiatrie
34295 Montpellier cedex 5
Ophtalmologie pédiatrique pour le dépistage et la surveillance des troubles de la vue - Centre de référence
Hôpital Gui de Chauliac - CHRU de Montpellier
Département d'ophtalmologie
34295 Montpellier cedex 5
Psychiatrie de l'enfant et de l'adolescent en cas de troubles comportementaux - Centre de compétence - Centre du secteur territorial en première intention
SMPEA Peyre Plantade - CHRU de Montpellier
34295 Montpellier cedex 5
Urologie pédiatrique pour les problèmes des voies génito-urinaires - Centre de compétence
Hôpital Lapeyronie - CHRU de Montpellier
Pôle enfant - Département de chirurgie infantile
34295 Montpellier cedex 5