Guide pratique : Syndrome de Waardenburg
Qu'est-ce que le syndrome de Waardenburg ?
Guide rédigé par Dr Camille Lemattre et Dr Patricia Blanchet Centre de référence Anomalies du développement CHU MONTPELLIER
Dernière mise à jour février 2018
Le syndrome de Waardenburg est une maladie génétique décrite pour la première fois en 1951, associant, à des degrés variables d’une personne à l’autre :
- des anomalies de la pigmentation de la peau, des cheveux ou de l’iris,
- une surdité,
- des particularités morphologiques faciales.
Il en existe quatre sous-types secondaires à des mutations distinctes (cf Diagnostic génétique et moléculaire).
Ces pathologies, résultant d’anomalies de la migration des cellules de la crête neurale lors du développement embryonnaire, sont regroupées sous le terme de neurocristopathies.
L’expressivité des signes est très variable, mais un sujet atteint du syndrome de Waardenburg exprime presque toujours au moins un symptôme (pénétrance quasi complète).
Le
Le diagnostic génétique est important à poser car le dépistage et la prise en charge de la surdité sont primordiaux dès les premiers mois de vie afin de permettre un développement optimal du langage oral et ainsi prévenir les complications.
Le
Diagnostic génétique et moléculaire
Le diagnostic du syndrome de Waardenburg est avant tout clinique. Cependant, la confirmation moléculaire est utile pour le pronostic, notamment en cas de surdité, ainsi que pour le conseil génétique à donner aux parents et au patient à l’âge adulte.
De plus, en fonction du gène impliqué, certains symptômes cliniques spécifiques pourront être recherchés. Le diagnostic moléculaire peut être obtenu par la recherche d’une anomalie dans l’un des gènes actuellement connus comme responsables de ces syndromes.
- Le syndrome de Waardenburg de type I (WS1, OMIM #193500) est caractérisé par une anomalie dans le gène PAX3 (OMIM *606597), à l’état hétérozygote. C’est-à-dire que, pour un sujet atteint, il suffit d’une mutation sur une de ses deux copies du gène PAX3 (soit celle héritée de son père, soit celle héritée de sa mère) pour être malade. Plus de 40 mutations dans PAX3 ont été décrites à ce jour. Toutefois, on ne retrouve pas de mutation dans ce gène chez environ 20% des patients atteints de WS1.
Pour les autres types, l’identification d’une mutation est plus rare.
- Le syndrome de Waardenburg de type III (WS3, OMIM #148820) ou syndrome de Klein-Waardenburg, est lui aussi causé par une anomalie du gène PAX3. Plusieurs mécanismes ont été identifiés. Il peut être lié à des mutations de PAX3 à l’état homozygote, c’est à dire présentant une mutation sur chacune des deux copies du gène. Une délétion (ou perte) de la totalité de ce gène, emportant parfois d’autres gènes situés à proximité, ou une mutation ponctuelle de PAX3, peut être responsable, à l’état hétérozygote. Ainsi, la présence d’une mutation de PAX3 à l’état hétérozygote est associée au syndrome de Waardenburg sans pour autant prédire la survenue du type I ou du type III.
- Le syndrome de Waardenburg de type II (WS2) présente une hétérogénéité génétique (plusieurs gènes peuvent être responsables). On distingue plusieurs sous-types. Une mutation à l’état hétérozygote dans le gène MITF (OMIM *156845) est identifiée dans 10 à 15% des cas et permet de définir le sous-type A (OMIM #193510). Une délétion à l’état homozygote du gène SNAI2 (OMIM *602150) permet de définir le sous-type D (OMIM #608890). Le sous-type E (OMIM #611584) est dû à une mutation à l’état hétérozygote du gène SOX10 (OMIM *602229). Aucun gène n’a encore été identifié dans les sous-types B et C. Récemment, les gènes KITLG (OMIM *184745) et EDNRB (OMIM *131244) ont été mis en cause dans WS2. Dans la majorité des cas, le gène en cause n’est cependant pas connu.
- Trois gènes différents sont responsables du syndrome de Waardenburg de type IV (WS4) ou syndrome de Shah-Waardenburg. Il s’agit de mutations à l’état homozygote dans les gènes EDNRB (sous-type A, OMIM #277580) et EDN3 (OMIM *131242) (sous-type B, OMIM #613265) et à l’état hétérozygote dans le gène SOX10 (sous-type C, OMIM #613266). 20 à 30% des patients WS4 ne sont pas porteurs de mutation dans ces trois gènes.
|
Type |
Description Initiale |
Transmission |
Génétique |
|
I |
Cf Principaux symptômes |
AD |
80% de mutations PAX3 (HTZ) |
|
II |
Pas d’écartement trop important des canthi internes ± forme neurologique |
AD
AR AD AD AD |
10 à 15% de mutations MITF (HTZ) délétion de SNAI2 (HMZ) SOX10 (HTZ) KITLG (HTZ) EDNRB (HTZ) |
|
III (Klein-Waardenburg) |
Anomalies des membres |
AD ou AR |
Mutations PAX3 (HMZ ou HTZ) (88%) ou délétion large emportant PAX3 (HTZ) (12%) |
|
IV (Shah-Waardenburg) |
Maladie de Hirschsprung ± forme neurologique |
AD
AR |
SOX10 (HTZ) (>50%) Mutations EDN3 ou EDNRB (HMZ) (20-30%) |
-
 Des anomalies spécifiques du gène SOX10 (large délétion ou mutation localisée au niveau des deux dernières régions codantes du gène) semblent conduire à une forme neurologique associant une maladie démyélinisante périphérique et centrale au syndrome de Waardenburg.
Des anomalies spécifiques du gène SOX10 (large délétion ou mutation localisée au niveau des deux dernières régions codantes du gène) semblent conduire à une forme neurologique associant une maladie démyélinisante périphérique et centrale au syndrome de Waardenburg.
AD : autosomique dominant ; AR : autosomique récessif (cf Conseil génétique)
HTZ : hétérozygote (une seule copie du gène concernée) ; HMZ : homozygote (deux copies du gène concernées)
Une mutation correspond à une anomalie visualisée dans un gène, pouvant être responsable de perturbation dans le fonctionnement de ce gène et donc d’une maladie.
Principaux symptômes
Le
Symptômes communs :
- Une surdité de perception congénitale, uni ou bilatérale et de degré variable. 71% des patients atteints d’un syndrome de Waardenburg ont une surdité de perception bilatérale. On la retrouve plus fréquemment dans WS2 (92% des cas) que dans les autres sous-types (52% pour WS1, 57% pour WS3 et 83,5% pour WS4), et également plus fréquemment dans le cadre de mutations des gènes SNAI2 (100%), SOX10 (96,5%) et MITF (90%). Elle est classiquement non évolutive, sauf dans le cadre de WS2 (évolutive dans 70% des cas).
- Des anomalies pigmentaires oculaires (dans 40% des cas) : hétérochromie irienne (iris présentant deux couleurs différentes pour un même œil), totale ou en secteur, uni ou bilatérale, parfois symétrique ; deux iris de couleurs différentes (yeux vairon) ; deux iris bleu pâle ou bleu saphir ; aspect de « fond d'œil albinoïde » (visualisation très nette des vaisseaux de la choroïde).
- Des anomalies des phanères (cheveux et poils) : une mèche blanche frontale dans les cheveux pouvant aller d'une touffe épaisse à quelques rares cheveux blancs (45% des cas). Cette mèche blanche peut se situer n'importe où sur la tête et même parfois disparaître. Un blanchiment prématuré des cheveux (canitie) est considéré comme un équivalent de la mèche blanche (30% des cas). Une décoloration des poils est possible lorsqu’ils sont localisés au niveau de tâches cutanées hypopigmentées (cf Anomalies dermatologiques).
- Des anomalies dermatologiques (28% des cas) : zones cutanées non ou peu pigmentées uniformes ou parsemées d'îlots hyperpigmentés. L'étendue de ces zones dépigmentées est variable.
- Des troubles de la perméabilité des voies lacrymales, responsables de larmoiement.
- Un élargissement de la base du nez, dans 70% des cas.
- Une hypertrichose sourcilière, prédominant dans la partie interne des sourcils, voire un synophris (confluence des sourcils sur la ligne médiane) et s'accompagnant parfois d'une décoloration des sourcils dans leur partie la plus interne.
- Une anomalie des paupières au niveau de leur jonction interne (canthi internes), rendant non visible ou peu visible le « blanc » de l’œil, dans sa partie proche du nez (dystopie des canthi internes), présente dans 68% des cas.
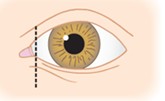
Œil normal
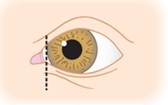
Dystopie des canthi internes
- Des anomalies plus rares, comme des anomalies de la vision (amblyopie, strabisme), une fente palatine ou labiale, un spina bifida (non fermeture du rachis autour de la moelle épinière) ou des symptômes vestibulaires (vertiges, troubles de l’équilibre) peuvent se rencontrer.
Symptômes spécifiques à WS3 :
- Des anomalies des membres : système musculaire peu développé (hypoplasique), contractures en flexion, fusion des os du carpe (os du poignet), syndactylies (fusion complète ou partielle de doigts ou orteils). Cependant, les syndactylies partielles des 2e et 3e orteils sont très fréquentes dans la population générale.
Symptômes spécifiques à WS4 :
- Anomalies abdominales : maladie de Hirschsprung (anomalie de fonctionnement de la partie terminale de l’intestin se traduisant par une constipation ou une occlusion intestinale, résultant de l’absence de développement congénital des cellules neuroganglionnaires permettant la régulation du péristaltisme intestinal).
Symptômes spécifiques aux mutations du gène SOX10 :
- Anomalies neurologiques : neuropathie périphérique, déficience intellectuelle de sévérité variable, ataxie cérébelleuse (instabilité debout immobile et pendant la marche), spasticité (augmentation anormale du tonus musculaire), hypotonie néonatale, épilepsie, nystagmus (mouvement d'oscillation involontaire et saccadée du globe oculaire). Une dysautonomie (productions de salive, de larmes et de sueur réduites, bradycardies et troubles du rythme cardiaque ou arythmies) peut également apparaître.
Diagnostic
Il est exceptionnel de présenter la symptomatologie complète. Il est admis une liste de critères majeurs et mineurs des syndromes de Waardenburg de type I et II.
Le diagnostic de WS1 et WS2 est posé si un sujet est porteur d'au moins deux signes majeurs ou d'un seul signe majeur associé à au moins deux signes mineurs.
|
Critères majeurs |
|
|
Critères mineurs |
|
Pour WS2, le blanchiment prématuré des cheveux est un critère majeur de diagnostic. Les symptômes neurologiques, dont une déficience intellectuelle, peuvent être présents si le gène SOX10 est en cause.
Le
Le
Conseil génétique
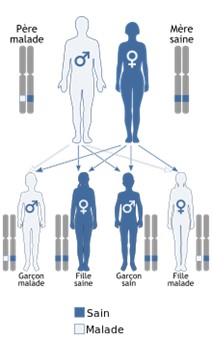
Le mode de transmission des syndromes de Waardenburg, liés aux gènes PAX3, MITF, SOX10, KITLG ainsi qu’à certaines mutations d’EDNRB, est autosomique dominant. Cela signifie que les personnes atteintes peuvent aussi bien être des femmes que des hommes, et qu’elles ont 50% de risques de transmettre l’anomalie à leurs enfants, quel que soit le sexe, et donc 50% de chances de ne pas la leur transmettre.
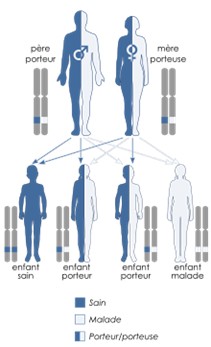
Le mode de transmission des syndromes de Waardenburg, liés aux gènes SNAI2, EDN3, à certaines mutations d’EDNRB, ainsi que celui de certaines formes de WS3 liées au gène PAX3, est autosomique récessif. De la même façon, les patients atteints peuvent être des femmes comme des hommes. Cependant, dans la transmission récessive, une mutation sur chacune des deux copies du gène est nécessaire pour être malade. Les parents sont porteurs d’une seule mutation et non malades car ils possèdent une copie normale du gène. Ils sont donc porteurs hétérozygotes. Pour chaque future grossesse, le risque d’avoir un autre enfant malade est de 25% (soit ¼).
Transmission autosomique récessive Transmission autosomique dominante
Prise en charge médicale initiale
Au moment du diagnostic, l’évaluation initiale cherche à faire le point sur les problèmes médicaux à prendre en charge. Elle est le plus souvent coordonnée par un généticien clinicien ou un pédiatre du Centre de Référence « Anomalies du Développement et Syndromes Malformatifs » en relation avec le pédiatre ou le médecin traitant du patient. Selon les complications associées, d’autres spécialistes pourront intervenir dans le suivi.
Bilan clinique
- pour évaluation des symptômes digestifs), conseil génétique et orientation vers spécialiste.
- Consultation dermatologique initiale complète pour évaluation des anomalies cutanées et des phanères et photographies éventuelles.
- Consultation ORL initiale pour évaluation de l’audition.
- Consultation gastrologique si anomalies digestives.
- Consultation neurologique si anomalies neurologiques.
- Consultation orthopédique si anomalies squelettiques.
- Consultation cardiologique si présence de signes de dysautonomie.
Bilan complémentaire systématique
- Fond d’œil à la recherche d’un aspect albinoïde ou d’anomalies de la pigmentation.
- Audiogramme pour dépistage et évaluation de la surdité.
Bilan complémentaire spécialisé dirigé par la clinique
- Radiographies du squelette à la recherche des critères diagnostiques du WS3.
- En cas de suspicion de maladie de Hirschsprung, une rectoscopie pour biopsie peut être envisagée.
- IRM cérébrale et électroencéphalogramme selon la clinique.
- Electrocardiogramme ou Holter cardiaque.
Suivi (cf Calendrier de suivi)
La prise en charge des personnes atteintes du syndrome de Waardenburg repose sur différentes approches.
Une consultation ORL annuelle est nécessaire pour le dépistage et la prise en charge des anomalies auditives, dès le plus jeune âge, afin de permettre un développement normal du langage oral. En cas de surdité, un appareil auditif ou un implant cochléaire permettra d'améliorer l'ouïe. L’implant cochléaire, dans cette indication, a de très bons résultats et permet l’acquisition du langage oral s’il est mis en place dès les premiers mois de vie.
Le suivi dermatologique se fait par une consultation dermatologique annuelle permettant une évaluation des anomalies cutanées afin de dépister l’apparition éventuelle de cancers cutanés. Une protection solaire rigoureuse est indispensable : ces personnes sont très sensibles au rayonnement UV car elles produisent peu de mélanine, le pigment protecteur de la peau. Ainsi, il est nécessaire d'appliquer une crème solaire et de porter des vêtements couvrants. Le port de lunettes de soleil est également nécessaire.
En fonction de la symptomatologie spécifique de chaque patient, d’autres consultations de suivi pourront être proposées, notamment des consultations de gastrologie, d’orthopédie, de neurologie ou de cardiologie.
Une intervention chirurgicale, à visée esthétique, pour corriger la dystopie des canthi internes, peut parfois être envisagée.
En raison de l’augmentation du risque d’anomalie de fermeture du tube neural tel que le spina bifida, une supplémentation en acide folique (Vitamine B9) est recommandée au moins deux mois avant la fécondation et pendant le premier trimestre de la grossesse à la dose de 5mg par jour. Cette prévention diminue de manière significative ce risque.
Associations de patients
ARIEDA - Association Régionale pour l’Intégration des Enfants Déficients Auditifs : www.arieda.fr/
CESDA34 – Centre d’Education Spécialisée pour Déficients Auditifs : www.cesda34.org/
ANPEDA - Association Nationale de Parents d'Enfants Déficients Auditifs : ANPEDA
ANPSA - Association Nationale pour les Personnes SourdAveugles : www.anpsa.fr/
Références
Waardenburg P. J. « A new syndrome combining developmental anomalies of the eyelids, eyebrows and noseroot with pigmentary anomalies of the iris and head hair and with congenital deafness; Dystopia canthi medialis et punctorum lacrimalium lateroversa, hyperplasia supercilii medialis et radicis nasi, heterochromia iridum totaliis sive partialis, albinismus circumscriptus (leucismus, polioss) et surditas congenita (surdimutitas) ». American Journal of Human Genetics 3, no 3 (septembre 1951): 195‑253.
Issa Sarah, Bondurand Nadège, Faubert Emmanuelle, Poisson Sylvain, Lecerf Laure, Nitschke Patrick, Deggouj Naima, Loundon Natalie, Jonard Laurence, David Albert, Sznajer Yves, Blanchet Patricia, Marlin Sandrine, Pingault Véronique. « EDNRB mutations cause Waardenburg syndrome type II in the heterozygous state ». Human Mutation. 2017;38:581–593.
Milunsky Jeff Mark. « Waardenburg Syndrome Type I ». In GeneReviews(®), édité par Roberta A. Pagon, Margaret P. Adam, Holly H. Ardinger, Stephanie E. Wallace, Anne Amemiya, Lora JH Bean, Thomas D. Bird, et al. Seattle (WA): University of Washington, Seattle, 1993. http://www.ncbi.nlm.nih.gov/books/NBK1531/.
Pingault Véronique, Dorothée Ente, Florence Dastot-Le Moal, Michel Goossens, Sandrine Marlin, et Nadège Bondurand. « Review and Update of Mutations Causing Waardenburg Syndrome ». Human Mutation 31, no 4 (1 avril 2010): 391‑406. doi:10.1002/humu.21211.
Drozniewska Malgorzata, et Olga Haus. « PAX3 gene deletion detected by microarray analysis in a girl with hearing loss ». Molecular Cytogenetics 7 (29 avril 2014): 30. doi:10.1186/1755-8166-7-30.
Ogawa Yasushi, Michihiro Kono, et Masashi Akiyama. « Pigmented Macules in Waardenburg Syndrome Type 2 Due to KITLG Mutation ». Pigment Cell & Melanoma Research, 2017, n/a-n/a. doi:10.1111/pcmr.12597.
Bogdanova-Mihaylova Petya, Michael D Alexander, Raymond PJ Murphy, et Sinéad M Murphy. « Waardenburg Syndrome: A Rare Cause of Inherited Neuropathy Due to SOX10 Mutation ». Journal of the Peripheral Nervous System, s. d., n/a-n/a. doi:10.1111/jns.12221.
Milunsky J. M., T. A. Maher, M. Ito, et A. Milunsky. « The Value of MLPA in Waardenburg Syndrome ». Genetic Testing 11, no 2 (2007): 179‑82. doi:10.1089/gte.2006.0531.
Bayrak Feda, Tolgahan Çatlı, Görkem Atsal, Taşkın Tokat, et Levent Olgun. « Waardenburg Syndrome: An Unusual Indication of Cochlear Implantation Experienced in 11 Patients ». The Journal of International Advanced Otology, 17 avril 2017. doi:10.5152/iao.2017.3017.
Nierop Josephine W.I. van, Rebecca R. Snabel, Margreet Langereis, Ronald J.E. Pennings, Ronald J.C. Admiraal, Emmanuel A.M. Mylanus, et Henricus P.M. Kunst. « Paediatric Cochlear Implantation in Patients with Waardenburg Syndrome ». Audiology & Neurotology 21, no 3 (juillet 2016): 187‑94. doi:10.1159/000444120
« Orphanet: Syndrome de Waardenburg ».
« Orphanet: Syndrome de Waardenburg Shah ».
« Orphanet: Syndrome de Waardenburg Shah variante neurologique ». http://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=FR&data_id=17538&Disease_Disease_Search_diseaseGroup=waardenburg&Disease_Disease_Search_diseaseType=Pat&Maladie(s)/groupes%20de%20maladies=Syndrome-de-Waardenburg-Shah-variante-neurologique&title=Syndrome-de-Waardenburg-Shah-variante-neurologique&search=Disease_Search_Simple.
« Orphanet: Syndrome de Waardenburg type 1 ».
« Orphanet: Syndrome de Waardenburg type 2 ».
« Orphanet: Syndrome de Waardenburg type 3 ».
Professionnels de santé en Occitanie
Centre de Référence des Anomalies du Développement et Syndromes Malformatifs : pour le diagnostic et la coordination des prises en charge à tout âge de la vie.
Hôpital Arnaud de Villeneuve
Pôle Biologie Pathologie - Département de Génétique Médicale
34295 Montpellier cedex 5
Centre de Compétence des Anomalies du Développement et Syndromes Malformatifs du sud-ouest Occitanie, Réunion
CHU de Nîmes - Groupe Hospitalo-Universitaire Caremeau
Laboratoire de Cytologie Clinique et Cytogénétique
30029 Nîmes cedex 9
Centre de Référence des Maladies Sensorielles Génétiques (MAOLYA) : pour le dépistage, la prise en charge et le suivi ORL et Ophtalmologique.
Hôpital Gui de Chauliac
Pôle Neurosciences Tête et Cou - Département d’Ophtalmologie
34295 Montpellier cedex 5
ORL pédiatrique : pour le dépistage et la surveillance des troubles de l’audition
Hôpital Gui de Chauliac
Pôle Neurosciences Tête et Cou - Département d’ORL et de Chirurgie cervico-faciale
34295 Montpellier cedex 5
ORL adulte : pour le suivi auditif à l’âge adulte
Hôpital Gui de Chauliac
Pôle Neurosciences Tête et Cou - Département d’ORL et de Chirurgie cervico-faciale
34295 Montpellier cedex 5
Dermatologie : pour la prise en charge des complications dermatologiques
Hôpital Saint-Eloi
Pôle Cliniques Médicales - Département de Dermatologie
34295 Montpellier cedex 5
Hépato-Gastro-Entérologie pédiatrique : pour la prise en charge des complications digestives et notamment de la maladie de Hirschsprung
Hôpital Arnaud de Villeneuve
Pôle Femme Mère Enfant - Département de Pédiatrie
34295 Montpellier cedex 5
Gastrologie adulte : pour le suivi à l’âge adulte
Hôpital Saint-Eloi
Pôle Digestif - Département d’Hépato-gastro-entérologie
34295 Montpellier cedex 5
Orthopédie pédiatrique : pour la prise en charge des complications du syndrome de Waardenburg de type III
Hôpital Lapeyronie
Pôle Femme Mère Enfant - Département de Chirurgie Infantile
34295 Montpellier cedex 5
Orthopédie adulte : pour le suivi à l’âge adulte
Hôpital Lapeyronie
Pôle Os et Articulations - Département de Chirurgie orthopédique et de Traumatologie
34295 Montpellier cedex 5
Neurologie pédiatrique : pour la prise en charge des complications neurologiques chez l’enfant
Hôpital Gui de Chauliac
Pôle Femme Mère Enfant - Département de Neuropédiatrie
34295 Montpellier cedex 5
Neurologie adulte : pour la prise en charge des complications neurologiques chez l’adulte
Hôpital Gui de Chauliac
Pôle Neuroscience Tête et Cou - Département de Neurologie
34295 Montpellier cedex 5
Cardiologie pédiatrique : pour la prise en charge des troubles du rythme cardiaque
Hôpital Arnaud de Villeneuve
Pôle Femme Mère Enfant - Département de Pédiatrie
34295 Montpellier cedex 5
Cardiologie adulte : pour le suivi à l’âge adulte
Hôpital Arnaud de Villeneuve
Pôle Cœur Poumons - Département de Cardiologie et Maladie Vasculaire
34295 Montpellier cedex 5