Guide pratique : Achondroplasie
Qu'est-ce que l'achondroplasie ?
Rédigé par Dr Flavien ROUXEL, Dr Marjolaine WILLEMS, responsable Centre de Référence Maladies Osseuses Constitutionnelles (MOC)
Rédigé 23 Avril 2024 - Réf. N°OMIM 100800 N°Orphanet ORPHA:15
L’achondroplasie est une maladie constitutionnelle des os, dont le gène responsable a été découvert en 1994. Il est dû à une variation pathogène activatrice au niveau d’un gène appelé FGFR3, situé au niveau du bras court du chromosome 4 en situation 4p16.3.
C’est un syndrome d’expression autosomique dominante, ce qui veut dire qu’il est lié à une atteinte d’un autosome, non lié au sexe, et qu’il suffit d’un exemplaire muté du gène pour être malade (on parle alors d’hétérozygotie).
Il entraine principalement un nanisme par atteinte rhizomélique, c’est-à-dire touchant la racine des membres supérieurs et inférieurs, avec une taille adulte moyenne de 131cm pour les hommes et 124cm pour les femmes, associé à un tronc long et étroit et des particularités de croissance des os du visage incluant une macrocéphalie avec front bombé et une hypoplasie médiofaciale. Les autres problèmes fréquents sont une hypotonie, un foramen magnum de petite taille ou de forme anormale pouvant entrainer une compression médullaire haute, un syndrome d’apnées obstructives du sommeil dû à l’hypoplasie médiofaciale, un retard d’acquisition de la motricité globale et un surpoids ou obésité, et, à l’âge adulte, une sténose rachidienne lombaire et des complications cardiovasculaires liées à l’obésité et aux apnées du sommeil si celles-ci ne sont pas prises en charge.
 Ci-contre, un chromosome 4. Le gène FGFR3, responsable d’achondroplasie lorsqu’il présente un variant pathogène (appelé anciennement « mutation »), se trouve sur la bande 16.3
Ci-contre, un chromosome 4. Le gène FGFR3, responsable d’achondroplasie lorsqu’il présente un variant pathogène (appelé anciennement « mutation »), se trouve sur la bande 16.3
Caractéristiques cliniques
L’achondroplasie étant un syndrome rare, il faut noter que la description clinique donnée ci-après représente les personnes déjà décrites dans la littérature, mais que celle-ci peut évoluer avec l’ajout de nouvelles connaissances sur l’achondroplasie.
Ce syndrome associe :
Sur le plan musculosquelettique :
- Petite taille disproportionnée : Le diagnostic d’achondroplasie est le plus souvent suspecté après 28 semaines d’aménorrhée, devant la présence d’os longs courts car le raccourcissement des os n’est habituellement pas encore présent à un terme plus précoce.
Un signe anténatal plus précoce d’achondroplasie est l’élargissement de l’angle fémoral diaphyso-métaphysaire, avec un angle augmenté de 36% chez les fœtus présentant une achondroplasie en comparaison aux fœtus non atteints (médiane de 125 degrés chez les fœtus atteints vs 95 degrés chez les fœtus non atteints). Le risque relatif d’achondroplasie est de 18.2 si l’angle est >108.5 degrés, et de 164 si l’angle est >116 degrés ; la mesure peut inconstamment être prise au deuxième trimestre, ce qui en fait un marqueur précoce de détection de l’achondroplasie.
La taille à la naissance des patients présentant une achondroplasie se situe à -2.9 déviations standard, avec une diminution lors des 3 premières années de vie pour atteindre un rythme de croissance situé sur la courbe des -5 déviations standard jusqu’à l’adolescence.
La taille adulte moyenne est de 131+/- 5.6cm pour les hommes et 124+/- 5.9cm pour les femmes.
- Macrocéphalie : La majorité des enfants atteints d’achondroplasie présente une macrocéphalie, et une petite proportion (environ 5%) présentera une hydrocéphalie nécessitant un traitement.
- Jonction craniocervicale étroite : historiquement, il existait une surmortalité précoce par complications liées à une compression de la moelle au niveau de la jonction craniocervicale, entrainant des apnées centrales. Ce risque peut être prévenu par l’examen neurologique complet des enfants, et la réalisation systématique à 3 mois de vie +/- répété avant l’âge de 12 mois :
- Une IRM de la charnière cervico-occipitale à la recherche d’une sténose du foramen magnum et d’une polysomnographie.
- Une intervention neurochirurgicale est envisagée pour les sténoses cliniquement significatives.
- Cyphose de la jonction thoracolombaire : présente chez 90-95% des petits enfants présentant une achondroplasie. Dans 10% des cas, elle ne se résout pas spontanément et peut entrainer des séquelles neurologiques, esthétiques, et fonctionnelles. Elle est prévenue par des mesures posturales et la kinésithérapie, et traitée par kinésithérapie et mise en place précoce d’un corset, avec ajustement régulier.
- Sténose spinale : La sténose spinale de L1-L4 est la cause la plus fréquente de recours médical chez l’individu adulte atteint d’achondroplasie, avec des symptômes allant de la claudication intermittente à l’exercice à l’altération permanente de fonction du membre inférieur ou à l’incontinence. Elle peut survenir dès l’adolescence et indique un interrogatoire et un examen neurologique systématique lors de chaque consultation de suivi afin de la dépister. Le traitement des formes symptomatiques est neurochirurgical.
- Hyperlaxité ligamentaire : Les individus atteints d’achondroplasie présentent de plus une hyperlaxité ligamentaire, généralement sans conséquence hormis un décalage des apprentissages moteurs. Dans de rares cas, la mise en place d’orthèses, notamment au niveau des genoux, peut être temporairement indiquée.
- Genu varum : il s’agit d’une déformation des membres inférieurs, complication très fréquente durant l’enfance, pour laquelle une prise en charge chirurgicale peut être indiquée.
Sur le plan endocrinologique :
L’obésité est un problème majeur dans l’achondroplasie, avec un gain excessif de poids pouvant se manifester dès la petite enfance. Chez l’adulte, l‘obésité peut aggraver la morbidité liée à la sténose lombaire et augmenter la mortalité par causes cardiovasculaires.
Une prévention et une prise en charge précoces sont indiquées avec mesures hygiénodiététiques.
Sur le plan neurologique et du développement :
Une hypotonie légère à modérée est classiquement présente durant l’enfance. Les enfants atteints d’achondroplasie ont du mal à tenir leur tête à cause de l’hypotonie et de la macrocéphalie. Une hypermobilité des petites articulations et des doigts courts peuvent retarder le développement moteur. Le développement du langage peut être retardé suite à une surdité de transmission. La mise en place de kinésithérapie précoce puis de psychomotricité sont indiquées pour soutenir la mise en place des étapes du développement.
Sur le plan psychosocial et cognitif :
L’intelligence est habituellement normale sauf en cas d’hydrocéphalie ou d’autres complications liées au système nerveux central. Une étude menée par Wigg et al. en 2016 sur 14 individus atteints d’achondroplasie âgés de 6 à 15 ans a montré un QI moyen de 92, avec une diminution significative dans les domaines du vocabulaire, du raisonnement verbal abstrait et de la mémoire de travail, ainsi qu’au niveau des fonctions exécutives. Les individus présentant une achondroplasie présentaient de plus des valeurs diminuées sur tous les tests d’attention par rapport aux individus non atteints. Les scores concernant le vocabulaire, la mémoire de travail et les compétences arithmétiques étaient quant à eux similaires aux enfants non atteints du même âge. Il est à souligner que ces difficultés sont inconstantes mais peuvent nécessiter une prise en charge rééducative lorsqu’elles sont présentes
Sur le plan pneumologique :
Durant l’enfance une partie des individus atteints d’achondroplasie présenteront une maladie pulmonaire restrictive, due à la présence d’un thorax petit et d’une compliance de la cage thoracique plus élevée, ce qui peut amener à la présence d’une hypoxémie chronique.
Les apnées du sommeil obstructives sont fréquentes chez les individus présentant une achondroplasie, chez les enfants comme chez les adultes. Les signes cliniques pouvant faire évoquer des apnées du sommeil sont : difficultés à l’éveil, somnolence diurne excessive, pauses respiratoires durant le sommeil, ronflements, difficultés de concentration durant la journée, irritabilité, dépression, hypertension artérielle.
Sur le plan ORL, maxillo-facial et dentaire :
- Apnées du sommeil : Il existe un risque important de syndrome d’apnée du sommeil, qui peut survenir tout au long de la vie des patients avec achondroplasie. La prévalence varie selon les études (de 10 à 80 %), mais les valeurs hautes (50 à 80%) semblent être particulièrement pertinentes, que ce soit chez l’enfant ou l’adulte.
Les apnées du sommeil peuvent être d’origine obstructives, centrales ou les deux. Les apnées centrales se rencontrent plus fréquemment dans les premières années de vie, en lien avec une compression de la moelle épinière au niveau du trou occipital (foramen magnum). Les apnées obstructives peuvent se rencontrer chez l’enfant comme chez l’adulte. Leur étiologie peut résulter d’une combinaison de facteurs : hypoplasie de l’étage moyen, fosses nasales étroites, hypertrophie adénoïdienne (souvent à partir de 2 ans) ou amygdalienne (débutant habituellement plus tard que l’hypertrophie adénoïdienne), macroglossie relative, hypotonie des muscles pharyngés.
Un syndrome d’apnée du sommeil doit être recherché et évalué dans l’idéal par polysomnographie, du fait de la coexistence possible d’apnées obstructives et centrales. Celle-ci doit être demandée au moment du diagnostic (idéalement avant 6 mois en cas de diagnostic précoce) et en cas de signes cliniques évocateurs d’apnée du sommeil. Un examen ORL est également nécessaire en cas de signes cliniques ou en cas de syndrome d’apnées obstructives du sommeil révélé à la polysomnographie, en vue de proposer une prise en charge chirurgicale. Les gestes les plus fréquemment proposés sont l’adénoïdectomie, l’adénoïdo-amygdalectomie (enfant souvent plus grands), la turbinoplastie des cornets inférieurs (à tout âge). La forte incidence d’apnées résiduelles après chirurgie des voies aériennes supérieures justifie la réalisation d’un contrôle systématique par poly(somno)graphie 2 à 4 mois après la chirurgie. En cas d’apnées résiduelles une ventilation nocturne peut être proposée, ou un traitement orthodontique (expansion maxillaire). Le traitement orthodontique aura également comme objectif de normaliser l’articulé dentaire (classe III fréquente). Chez l’enfant plus grand, une fois la croissance osseuse maxillo-faciale terminée, une chirurgie orthognatique (ostéotomie de type Lefort I ou Lefort III) peut être nécessaire.
Outre les anomalies de l’articulé dentaire, les patients peuvent présenter un taurodontisme.
L’effet des nouveaux traitements (comme le vosoritide) sur la croissance maxillofaciale n’est pas encore bien connu.
Les enfants peuvent présenter un retard de langage qui peut nécessiter une rééducation orthophonique.
- Surdité : Les patients atteints d’achondroplasie peuvent également présenter des surdités et des problèmes d’otite chronique.
Les otites séromuqueuses sont fréquentes, prédominant chez l’enfant mais pouvant également être présentes chez l’adulte. Un suivi ORL régulier est nécessaire. En cas d’épanchement rétrotympanique associé à une perte auditive de plus de 25 dB, au vu du risque de retard de langage inhérent à l’achondroplasie, des aérateurs transtympaniques peuvent être proposés. Des procidences du golfe de la jugulaire ont été décrits de façon plus fréquente qu’en population générale, qui peuvent entraîner des surdités de transmission unilatérale. Il existe également un risque de saignement lors d’un geste chirurgical au niveau de l’oreille moyenne, ce que le chirurgien ORL doit garder en tête.
La surdité est plus fréquente chez les patients avec achondroplasie par rapport à la population générale, et ce quelle que soit la tranche d’âge. Elle peut donc être de transmission (en lien avec les problème d’otite chronique, de dysfonctionnement du tube auditif ou d’une malposition du golfe de la jugulaire) mais mixte ou neurosensorielle. Les mécanismes sous-jacents d’atteinte de l’oreille interne ne sont pas clairement décrits.
Risque anesthésique en lien avec la malformation craniofaciale :
Les patients avec achondroplasie peuvent également présenter des difficultés de ventilation voire d’intubation (petite ouverture de bouche, macroglossie relative, étroitesse des fosses nasales, hypoplasie de l’étage moyen, obstruction des voies aériennes par hypertrophie adénoïdo-amygdalienne). Chez l’enfant, en cas de sténose du foramen magnum, l’extension et mouvements cervicaux doivent être limités ou évités. Si le risque de sténose du foramen magnum n’est pas éliminé, ou si la sténose est avérée, l’utilisation de vidéo-laryngoscope peut être nécessaire pour limiter les mouvements cervicaux lors de l’intubation. Cet équipement peut également être utile en cas d’intubation difficile.
Aspects génétiques de l’achondroplasie
Nous possédons tous 46 chromosomes, répartis en 23 paires avec, pour chaque paire, un chromosome hérité de notre mère et un chromosome hérité de notre père. Parmi ces 23 paires de chromosomes, on distingue les autosomes (chromosomes 1 à 22) qui sont communs aux filles et aux garçons, et les gonosomes qui sont les chromosomes sexuels : XX chez les filles et XY chez les garçons. Chacun de ces chromosomes porte des centaines de gènes, qui permettent le bon fonctionnement de l’organisme grâce à la production de protéines.
Dans l’achondroplasie, une des copies du gène FGFR3 est atteinte et entraine une suractivation de FGFR3. Deux variations sont principalement responsables d’achondroplasie : c.1138G>A et c.1138G>C (la 1138e lettre du gène FGFR3, normalement un G, devient un A ou un C).
L’autre copie de FGFR3 est normale. On parle d’expression autosomique dominante.
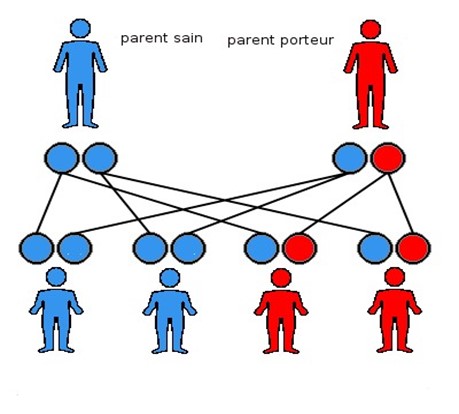
Lors de la conception d’un embryon, la maman donne 23 chromosomes, et le papa donne aussi 23 chromosomes, ce qui donne 4 combinaisons possibles de patrimoine génétique pour l’enfant, schématisées ci-dessus. Chaque rond représente un lot de chromosomes.
Dans le cadre d’une maladie autosomique dominante, le parent porteur a donc un risque sur deux de transmettre la mutation (ici schématisée en rouge). L’enfant a un risque sur deux de porter la variation pathogène, et donc de présenter la maladie.
Le diagnostic
Le diagnostic d’achondroplasie peut s’établir sur la base de l’analyse du tableau clinique et des radiographies. Les aspects radiographiques classiques d’achondroplasie sont : une forme carrée du pelvis avec une petite échancrure sacrosciatique, des pédicules vertébraux courts avec rapprochement interpédiculaire de la région thoracique basse à la région lombaire, un raccourcissement rhizomélique (de la racine des membres) des os longs, une radiotransparence fémorale proximale et une forme en chevron des épiphyses fémorales distales.
Dans le cas d’un tableau clinique et radiologique classique, il est possible de confirmer le diagnostic par analyse ciblée des variants pathogènes les plus communs : c.1138G>A et c.1138G>C.
Dans le cadre d’un doute diagnostic ou si le diagnostic n’a pas été identifié par le test de 1ère ligne, il est possible de réaliser l’analyse d’un panel de gènes incluant FGFR3 ainsi que d’autres gènes d’intérêt.
Le diagnostic génétique peut par ailleurs permettre de distinguer l’achondroplasie de différents diagnostics différentiels comme par exemple l’hypochondroplasie, causée par des variants pathogènes différents dans le gène FGFR3. Cette distinction est importante car elle peut avoir un retentissement sur le pronostic et sur le plan thérapeutique.
Conseils génétiques
Cas sporadique : Le cas de figure le plus fréquent (80% des cas) est la survenue de la pathologie chez un enfant de parents non atteints par la maladie. Il s’agit d’un cas sporadique. Dans ce cas, le variant pathogène dans le gène FGFR3 provient d’un « accident » génétique qui se produit dans le spermatozoïde ou l’ovule. On parle alors de variant pathogène (anciennement mutation) de novo.
Il peut arriver dans ce cas de figure que plusieurs spermatozoïdes ou plusieurs ovules portent le variant pathogène alors qu’il n’est présent nulle part ailleurs dans le corps des parents. Ce mécanisme s’appelle une mosaïque germinale.
Devant un risque de mosaïque germinale estimé à moins de 1%, une consultation de génétique est possible dans le but de proposer un diagnostic prénatal aux parents d’un enfant atteint pour une future grossesse s’ils désirent s’assurer que le fœtus ne porte pas le variant pathogène. Ce diagnostic prénatal n’est pas obligatoire et est à discuter avec la famille.
Il existe 2 façons de réaliser ce diagnostic prénatal :
- Le diagnostic prénatal invasif qui consiste en un prélèvement, soit par biopsie de trophoblaste à partir de 12 semaines d’aménorrhée, soit de liquide amniotique à partir de 15 semaines d’aménorrhée, afin de mettre en évidence le variant pathogène de FGFR3.
- Le diagnostic prénatal non invasif qui peut se réaliser à partir de 10 semaines d’aménorrhée en cas de variant pathogène de novo ou d’origine paternelle et consiste en une prise de sang de la femme enceinte, avec étude de l’ADN fœtal libre circulant.
Dans le cas d’un résultat mettant en évidence un variant pathogène de FGFR3 en période anténatale, il pourra être discuté, si la femme enceinte en fait la demande, la réalisation d’une interruption médicale de grossesse (IMG), sans limite de terme.
Cas hérité : Dans les 20% de cas restants, l’achondroplasie étant de transmission autosomique dominante, un individu atteint de la maladie a transmis sa maladie à ses enfants, avec un risque de transmission dans ce cas de 50% pour chaque enfant.
Dans ce cas de figure, il est possible de réaliser :
- Un diagnostic prénatal, invasif ou non invasif, comme décrit dans le paragraphe ci-dessus
- Un diagnostic préimplantatoire, ou DPI, qui consiste à réaliser une fécondation in vitro, un diagnostic génétique sur une à 2 cellules de l’embryon au stade très précoce, puis ne transférer dans l’utérus de la maman que les embryons indemnes. Il faut toutefois noter que le processus de DPI comprend plusieurs étapes incluant une stimulation ovarienne, le prélèvement des ovocytes, la réalisation d’une fécondation in vitro (FIV) puis l’analyse d’une à deux cellules de l’embryon, et enfin la sélection d’embryons non atteints avec transfert dans la cavité utérine. Le taux d’implantation global est compris entre 20 et 40 % par tentative, et le premier essai est réalisé au terme d’un parcours de soin d’une durée moyenne de 18 mois actuellement.
Pronostics
Une étude publiée en 2007 avec un suivi de près de 800 individus atteints d’achondroplasie a mis en évidence une mortalité augmentée, avec une espérance de vie semblant diminuée d’environ 10 ans. Cependant, avec l’amélioration de la prise en charge, la surmortalité de la petite enfance a été très largement diminuée, et le suivi multidisciplinaire tout au long de la vie est susceptible de modifier ces données.
Traitement
- Traitements chirurgicaux :
Il peut être nécessaire d’avoir recours à la chirurgie dans certains cas de complications de l’achondroplasie. Chez l’enfant, les chirurgies les plus fréquemment réalisées sont les interventions neurochirurgicales pour le traitement de la sténose du foramen magnum ou d’une éventuelle hydrocéphalie symptomatique, les chirurgies de l’espace ORL comme la myringotomie, la tonsillectomie ou encore l’adénoïdectomie, et plus rarement la chirurgie orthopédique pour correction de la cyphoscoliose thoracolombaire.
A l’âge adulte, la chirurgie la plus fréquente concerne le traitement de la sténose spinale lombaire.
Il est aussi possible d’avoir recours à la chirurgie orthopédique pour la correction du genu varum, ainsi que pour l’allongement des membres, avec des techniques chirurgicales variant suivant l’individu et suivant le centre pratiquant la chirurgie. Cette technique, lourde, est rarement envisagée par les patients du fait des complications, quasiment systématiques et des contraintes.
- Traitement médical :
Les traitements médicaux de l’achondroplasie incluent la prise en charge des apnées du sommeil par ventilation non invasive, le traitement de la cyphose thoracolombaire par corset anti-cyphotique et la prise en charge de l’obésité par des règles diététiques.
Il existe désormais un traitement permettant le ciblage et l’inhibition de la voie de signalisation MAPK, qui est suractivée chez les individus présentant une achondroplasie. Ce traitement, le vosoritide, est autorisé depuis 2021 en Europe pour le traitement des individus présentant une achondroplasie confirmée par test génétique, âgés de plus de 2 ans et jusqu’à fermeture des épiphyses, à administrer par injection quotidienne sous-cutanée. Dans un essai de phase 3 contrôlé randomisé en double aveugle, il a été montré sur un an une augmentation de la vitesse de croissance de 1.57cm/an et utilisant le vosoritide vs placebo. La poursuite sur une année supplémentaire de l’étude a montré une conservation de l’effet du médicament sur la seconde année de traitement, avec un gain de taille sur une période de 2 ans de 3.52cm. Les effets indésirables les plus fréquents incluent des réactions du site d’injection modérées, des vomissements et des diminutions transitoires de tension artérielle généralement asymptomatiques et de résolution spontanée, pouvant être prévenues par des précautions d’utilisation. Le vosoritide est de plus actuellement en cours d’évaluation pour les enfants de moins de 2 ans, et une étude de phase 2 récente a montré un profil d’évènement indésirables peu sévères, avec un gain de taille dans cette population. Si ces données venaient à être confirmées, le vosoritide pourrait prochainement être débuté à un âge encore plus précoce qu’actuellement.
De nombreuses molécules sont de plus actuellement en études de phase 2/3 dans l’achondroplasie, le traitement décrit ci-dessus sera donc potentiellement amené à changer dans les prochaines années. Nous manquons toujours de données concernant la sécurité d’utilisation à long terme de ces traitements.
Surveillance et suivi
L’achondroplasie nécessite un suivi spécialisé tout au long de la vie. Ce suivi sera coordonné par le médecin du centre de référence ou de compétence, en lien avec le pédiatre ou le médecin généraliste, et adapté en fonction des problèmes médicaux spécifiques du patient.
Suivi paramédical
Des prises en charges rééducatives adaptées aux besoins de chaque personne peuvent aussi être mises en place si besoin pour faciliter les apprentissages, telles que, très précocement, la kinésithérapie a visée de renforcement musculaire, lutte contre la cyphose et le flessum de hanches, entretien des mobilités articulaires et aide à l’acquisition des différentes étapes du développement moteur. La mise en place de psychomotricité en relais ou complément de la kinésithérapie est très fréquente, adaptée à l’évolution et aux besoins de chaque enfant. Un bilan et une prise en charge en ergothérapie peut être indiquée afin d’aider à l’adaptation de l’environnement.
Certains enfants présentant un retard de langage peuvent également bénéficier de la mise en place d’orthophonie.
Accès aux droits
Des aides financières, telle que l’Allocation pour l’Education des Enfants en situation de Handicap (AEEH), peuvent également être mises en place pour compenser la prise en charge du handicap. L’AEEH est basée sur les constatations des professionnels médicaux et paramédicaux d’une part, et des aidants principaux d’autre part.
Le médecin référent se charge de remplir la demande de prise en charge à 100% au titre d’une Affection Longue Durée (ALD) hors liste. Il remplit également le certificat médical pour le dossier de la Maison Départementale de l’Autonomie (MDA) (anciennement appelée Maison Départementale les Personnes en situation de Handicap, MDPH).
A l’âge adulte, différentes aides sont disponibles comme l’allocation aux adultes handicapés (AAH), dont la demande peut être faite auprès de la MDA et attribuée par la commission des droits et de l’autonomie des personnes handicapées (CDAPH), ainsi que la prestation de compensation du handicap (PCH) qui est versée par le conseil départemental et permet de compenser les charges liées à un besoin d’aide humaine, d’aide technique mais aussi d’aménagement du logement ou du véhicule ou des surcouts liés aux transports, et des charges spécifiques ou exceptionnelles liées au handicap. Il est possible de cumuler ces aides avec MaPrimeAdapt’ qui est une aide financière mise en place depuis janvier 2024 permettant d’aider au financement de travaux d’adaptation du logement à la situation de handicap.
Pour l’accès à l’emploi, il est possible d’obtenir la reconnaissance de la qualité de travailleur handicapé (RQTH) qui permet de bénéficier de différentes mesures comme l’aménagement des horaires de travail, l’adaptation du poste de travail mais aussi de bénéficier de dispositifs dédiés à l’insertion professionnelle.
En cas de limitation importante de l’autonomie de marche, il est possible d’obtenir la carte mobilité inclusion (CMI) mention « stationnement » afin de pouvoir stationner gratuitement et sans limite de durée sur toutes les places de stationnement public sur la voirie en surface.
En cas de station debout pénible, il est possible d’obtenir la CMI « priorité » qui permet une priorité d’accès aux places assises dans les transports en commun, les espaces et salles d’attente, les établissements et les manifestations accueillant du public, ainsi qu’une priorité d’accès dans les files d’attente.
Enfin, en cas de perte d’autonomie importante, il est possible d’obtenir l’accès à la CMI « invalidité » qui offre les mêmes avantages que la CMI « priorité » avec en plus des réductions dans les transports et des avantages fiscaux.
Associations de malades
Il existe l’Association des personnes de petite taille, qui a des missions d’information et d’accompagnement, est un lieu d’échange et de partage.
Voici le lien vers leur site internet : https://www.appt.asso.fr/
Références
Gilligan LA, Calvo-Garcia MA, Weaver KN, Kline-Fath BM. Fetal magnetic resonance imaging of skeletal dysplasias. Pediatr Radiol. 2020 Feb;50(2):224-233. doi: 10.1007/s00247-019-04537-8. Epub 2019 Nov 27. PMID: 31776601.
Hoover-Fong J, Scott CI, Jones MC; COMMITTEE ON GENETICS. Health Supervision for People With Achondroplasia. Pediatrics. 2020 Jun;145(6):e20201010. doi: 10.1542/peds.2020-1010. PMID: 32457214.
Horton WA, Rotter JI, Rimoin DL et al. Standard growth curves for achondroplasia. J Pediatr. 1978 Sep;93(3):435-8. doi: 10.1016/s0022-3476(78)81152-4. PMID: 690757.
Ireland PJ, Donaghey S, McGill J et al. Development in children with achondroplasia: a prospective clinical cohort study. Dev Med Child Neurol. 2012;54:532–7.
Khalil A, Chaoui R, Lebek H, Esser T, Entezami M, Toms J, Thilaganathan B. Widening of the femoral diaphysis-metaphysis angle at 20-24 weeks: a marker for the detection of achondroplasia prior to the onset of skeletal shortening. Am J Obstet Gynecol. 2016 Feb;214(2):291-292. doi: 10.1016/j.ajog.2015.09.089. Epub 2015 Oct 9. PMID: 26450406.
Legare JM. Achondroplasia. 1998 Oct 12 [Updated 2023 May 11]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1152/
Mogayzel PJ Jr, Carroll JL, Loughlin GM et al. Sleep-disordered breathing in children with achondroplasia. J Pediatr. 1998;132:667–71.
Pauli RM. Achondroplasia: a comprehensive clinical review. Orphanet J Rare Dis. 2019 Jan 3;14(1):1. doi: 10.1186/s13023-018-0972-6. PMID: 30606190; PMCID: PMC6318916.
Pauli RM, Horton VK, Glinski LP et al. Prospective assessment of risks for cervicomedullary-junction compression in infants with achondroplasia. Am J Hum Genet. 1995;56:732–44.
Pauli RM, Breed A, Horton VK et al. Prevention of fixed, angular kyphosis in achondroplasia. J Pediatr Orthop. 1997;17:726–33
Savarirayan R, Tofts L, Irving M et al. Once-daily, subcutaneous vosoritide therapy in children with achondroplasia: a randomised, double-blind, phase 3, placebo-controlled, multicentre trial. Lancet. 2020 Sep 5;396(10252):684-692. doi: 10.1016/S0140-6736(20)31541-5. Erratum in: Lancet. 2020 Oct 10;396(10257):1070. PMID: 32891212.
Savarirayan R, Tofts L, Irving M et al. Safe and persistent growth-promoting effects of vosoritide in children with achondroplasia: 2-year results from an open-label, Phase 3 extension study. Genet Med. 2021;23:2443–7
Savarirayan R, Wilcox WR, Harmatz P et al. Vosoritide therapy in children with achondroplasia aged 3-59 months: a multinational, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Child Adolesc Health. 2024 Jan;8(1):40-50. doi: 10.1016/S2352-4642(23)00265-1. Epub 2023 Nov 18. PMID: 37984383. Copy
Savarirayan, R., Ireland, P., Irving, M et al. (2022). International Consensus Statement on the diagnosis, multidisciplinary management and lifelong care of individuals with achondroplasia. Nat Rev Endocrinol, 18(3), 173-189. doi:10.1038/s41574-021-00595-x
Savarirayan R, Irving M, Harmatz P et al. Growth parameters in children with achondroplasia: A 7-year, prospective, multinational, observational study. Genet Med. 2022 Dec;24(12):2444-2452. doi: 10.1016/j.gim.2022.08.015. Epub 2022 Sep 16. PMID: 36107167.
Tenconi, R., Khirani, S., Amaddeo, A., Michot, C., Baujat, G., Couloigner, V., . . . Fauroux, B. (2017). Sleep-disordered breathing and its management in children with achondroplasia. Am J Med Genet A, 173(4), 868-878. doi:10.1002/ajmg.a.38130
Tunkel D, Alade Y, Kerbavez R, et al. Hearing loss in skeletal dysplasia patients. Am J Med Genet A. 2012;158A:1551–5.
Vivanti AJ, Costa JM, Rosefort A et al. Optimal non-invasive diagnosis of fetal achondroplasia combining ultrasonography with circulating cell-free fetal DNA analysis. Ultrasound Obstet Gynecol. 2019 Jan;53(1):87-94. doi: 10.1002/uog.19018. Epub 2018 Nov 28. PMID: 29380944.
Waller DK, Correa A, Vo TM et al. The population-based prevalence of achondroplasia and thanatophoric dysplasia in selected regions of the US. Am J Med Genet A. 2008;146A:2385–9.
Wigg K, Tofts L, Benson S et al. The neuropsychological function of children with achondroplasia. Am J Med Genet A. 2016;170:2882–8.
Wynn J, King TM, Gambello MJ, et al. Mortality in achondroplasia study: a 42-year follow-up. Am J Med Genet A. 2007;143A:2502–11.
Professionnels de santé en Occitanie Ouest
Centre de compétence des maladies osseuses constitutionnelles
CHU de Toulouse - Hôpital des enfants
Service de Pédiatrie - Endocrinologie, génétique et gynécologie médicale
Pr Thomas EDOUARD
330 avenue de Grande Bretagne
31059 Toulouse Cedex 9
05 34 55 85 49
Professionnels de santé en Occitanie Est
Centre de Référence maladies osseuses constitutionnelles
Hôpital Arnaud de Villeneuve
Département de génétique médicale
Dr Willems Marjolaine
371 avenue 371 Avenue Doyen Gaston Giraud 34295 Montpellier Cedex 5
04 67 33 65 64
Cardiopédiatrie
Hôpital Arnaud de Villeneuve
Département de Pédiatrie (Pôle Enfant)
371, avenue Doyen Gaston Giraud 34295 MONTPELLIER cedex 5 - CHRU de Montpellier
04 67 33 66 43
Cardiologie adulte
Hôpital Arnaud de Villeneuve
Département de Cardiologie et Maladies Vasculaires
371, avenue Doyen Gaston Giraud 34295 MONTPELLIER cedex 5 - CHRU de Montpellier
04 67 33 61 89 ou 04 67 33 62 01
Chirurgie orthopédique et plastique infantile : pour les scolioses
Hôpital Lapeyronie
Département de chirurgie infantile (Pôle Enfant)
371, avenue doyen Gaston Giraud 34295 MONTPELLIER cedex 5 - CHRU de Montpellier
04 67 33 87 65
Endocrinologie pédiatrique : pour les troubles hormonaux
Hôpital Arnaud de Villeneuve
Département de Pédiatrie (Pôle Enfant)
371, avenue Doyen Gaston Giraud 34295 MONTPELLIER cedex 5 - CHRU de Montpellier
04 67 33 66 43
Neuropédiatrie et neurochirurgie pédiatrique : suivi développemental et des complications neurologiques
Hôpital Gui de Chauliac
Département de Pédiatrie
80, avenue Augustin Fliche 34295 Montpellier cedex 5 - CHRU de Montpellier
04 67 33 77 37
ORL pédiatrique : pour la surveillance des infections ORL et des troubles de l’audition
Hôpital Gui de Chauliac
ORL et chirurgie cervico-faciale
80, avenue Augustin Fliche 34295 Montpellier cedex 5 - CHRU de Montpellier
04 67 33 68 04
Pneumologie pédiatrique / Pôle Femme mère enfant – département de pédiatrie
CHU de Montpellier – Hôpital Arnaud de Villeneuve
Maladies respiratoires rares
Dr Marie-Catherine Renoux
371 Avenue Doyen Gaston Giraud 34295 Montpellier Cedex 5
07 88 01 44 37